
In the healthcare field, informed consent is many things: It’s a legal doctrine, an ethical obligation, and a standard of care. According to oral maxillofacial surgeon Mughda Patwardhan, D.D.S., M.D., informed consent is a practice more than anything.
“Informed consent is basically where the healthcare provider outlines the risks, the benefits, and any alternative treatments that are available for a specific disease or medical issue,” Patwardhan says. “It’s a collaborative process in which the patient and the healthcare provider make decisions together.”
You’ll find this type of collaboration anywhere patients are treated or studied, in every corner of the medical field: clinical practice, research, pharmaceutical development, medical device manufacturing, and more. In all these instances, informed consent stems from the same root, a principle first written into U.S. law in 1914’s landmark Schloendorff v. Society of New York Hospital.
“Every human being of adult years and sound mind has a right to determine what shall be done with his own body,” wrote Justice Benjamin Cardozo of the New York Court of Appeals. “And a surgeon who operates without his patient’s consent commits an assault for which he is liable in damages.”
In short, informed consent is the process of patients determining what can and cannot be done with their bodies. This guide will tell you everything you need to know about informed consent in the healthcare industry.
Chapter synopsis
- Introduction
- The elements of informed consent. What details should you cover when practicing informed consent? Find out here.
- Why informed consent is mandatory. There are serious legal and professional consequences for violating informed consent. Learn why physicians are so careful when it comes to this subject.
- Who needs informed consent? The healthcare industry is much larger than you might think. Does your company need a consistent informed consent practice?
- How to build an informed consent form. Find out what to include on an informed consent form, along with some tips that can simplify sharing and collection.
A quick, important note before we go further. Along with informed consent, you might hear a term that’s phonetically similar, but substantially very different: implied consent. Informed consent is a formal process, usually ending with a signed document. With implied consent, by contrast, the patient’s behavior tells the healthcare provider that they agree to treatment.
Implied consent is good enough for many of the common, low-risk tasks that doctors perform. For example, you don’t need to collect informed consent to take a patient’s blood pressure as part of a scheduled exam. The fact that the patient showed up, sat down, and presented their arm for the blood pressure cuff implies consent. (Read more about implied consent vs informed consent here.)


With that distinction out of the way, here’s everything you need to know about informed consent. Feel free to skip through the chapters for quick answers to specific questions, and be sure to bookmark this guide for future use. Informed consent is a complex topic, and few in the healthcare industry can afford to get it wrong.
Elements of informed consent
The practice of informed consent may be spoken, written, or — more likely — both. But it cannot lack certain information.
Informed consent must include enough detail for patients to make an educated decision about how they’d like to proceed with care. After all, informed consent is a key component of shared decision-making in healthcare, an approach that invites patients to participate in their care plans.
The point of shared decision-making is to keep the patient in control, and control requires certain facts. Specifically, according to the American Medical Association, physicians should include at least the following items in their informed consent practice:
- A diagnosis. What illness, injury, or condition is the physician treating?
- A description of the intervention. What’s the treatment plan? Does it require medication, surgery, physical therapy, etc.? Physicians should provide an explanation and the hoped-for goals of each intervention they’d like to try.
- Any available alternative treatments, including skipping treatment altogether.
- The risks and benefits of each prospective intervention.
- Side effects and other potentially negative aspects of the treatment (e.g., the medication is unusually expensive, or you can never eat cheese again after the surgery).
Physicians should provide all of this information in clear, everyday language. “When you’re speaking with a patient, they may not be familiar with medical jargon,” says Patwardhan. “It’s really important to communicate in a way that helps the patient understand what is happening and why, as well as the alternatives and potential risks and benefits of the procedure.”
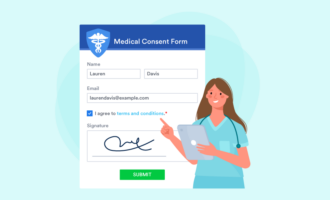
Healthcare providers must document their patient’s informed consent, which typically means having the patient sign a consent form. Signed consent forms should be included in the patient’s record.
Remember that documents containing personal information are subject to all the patient confidentiality protections afforded by the Health Insurance Portability and Accountability Act of 1996 (HIPAA). If you need electronic documentation at your fingertips, Jotform provides HIPAA-enabled online forms for medical use, including digital informed consent documentation with electronic signatures.
But before any documentation — before even beginning a conversation about informed consent — physicians must make sure the patient is healthy enough to understand. The technical term for this ability to process informed consent data is “decision-making capacity.”
Pro Tip
Use Jotform Sign Mobile to invite patients by link or email, capture in-person consent during visits, scan paper forms to e-sign, and track completion in Sign Inbox.
Medical decision-making capacity and informed consent
Decision-making capacity is a prerequisite for proper informed consent. If a patient can’t fully understand information about their treatment, they can’t agree to it — and if the patient is proven to have lacked decision-making capacity when they signed their consent form, that signature could be rendered legally invalid.
We’ll discuss the high costs of informed consent mistakes in Chapter 2. For now, suffice it to say that physicians must verify that patients have decision-making capacity before asking them to sign the paperwork.
The journal American Family Physician lists the four parts of decision-making capacity. Patients must be able to do the following:
- Show that they understand the proposed treatment, including alternatives, risks, and benefits.
- “Demonstrate appreciation” of the risks and benefits of treatments. It’s not enough to understand the details of an intervention. Patients with decision-making capacity have to connect those details to reality and show that they comprehend how their lives could change as a result of the treatment.
- Explain why they made a given choice. There must be some rational thinking leading to a patient’s decision.
- Communicate all of the above to the physician.
Decision-making capacity is usually easy to detect. Most patients have it, and physicians typically don’t need to conduct detailed evaluations before practicing informed consent. Sometimes, though, the doctor may need to issue a formal test for this capacity — if, for instance, the patient’s mental state changes suddenly.
Elements of informed consent in research
So far, we’ve discussed informed consent in the clinical healthcare field, that is, where patients are treated. (And yes, that includes mental health — read more about informed consent in psychology here.)
Informed consent is also crucial to the field of medical research: disease studies, drug trials, medical device testing, etc. The principles of informed consent in research match those of clinical medicine — the patient/subject must retain control over their participation.
This post covers informed consent in a research context. For a more detailed list of informed consent elements in the research industry, see the U.S. Office for Human Research Protections’s informed consent checklist.
The Office for Human Research Protection is part of the U.S. Department of Health and Human Services — which is just one of the many institutions that require medical professionals to practice informed consent with patients and subjects alike. There’s quite a bit of legal liability mixed in with informed consent practices. Keep reading to learn more about these risks.
Why is informed consent mandatory?
Informed consent is built into most doctor-patient interactions. It’s a routine part of every exam-room conversation and all pretreatment paperwork. But why?
Physicians care for their patients and want to practice shared decision-making. That’s the main reason informed consent remains nearly universal. But there are also outside forces that hold physicians accountable.
Physician behavior is tightly regulated by a web of accreditation organizations, medical associations, and laws. Here’s why it’s so important to establish and follow strong informed consent protocols at every healthcare facility.
What happens if informed consent is breached?
Different authorities have different powers of enforcement. Some can revoke medical licenses, while others may threaten funding. Either way, doctors who don’t comply with informed consent rules won’t practice long. Here are the top U.S. medical authorities that require informed consent, along with the consequences for physicians who don’t comply:
- The American Medical Association (AMA). The AMA Code of Medical Ethics, Opinion 2.1.1 holds that “patients have a right to receive information and ask questions about recommended treatments so that they can make well-considered decisions about care.” Specifically, the code states that physicians must check for decision-making capacity in their patients, present the elements of informed consent, and document the informed consent in the patient’s record. While the AMA doesn’t punish noncompliance itself, it does refer would-be reporters of unethical behavior to the relevant state medical licensing board. In extreme cases, these boards can revoke a physician’s license for violations of the AMA Code of Medical Ethics — including the informed consent requirement.
- The Centers for Medicare and Medicaid Services (CMS). Because it’s a federal agency, CMS statutes are written into law. The relevant legal code for informed consent is 42 CFR 482.24, which requires all medical practitioners who accept Medicare and Medicaid to record “properly executed informed consent forms for procedures and treatments specified by the medical staff,” including “written patient consent.” A healthcare provider who fails to comply with these codes may be cut off from CMS payments entirely. Medicare payments topped $731 billion in 2018, a significant amount that few hospitals could afford to lose.
- The Joint Commission. This authority provides accreditation for hospitals and other healthcare providers. To maintain Joint Commission accreditation, physicians must practice informed consent and submit proper medical documentation, including signed consent forms from all patients. Failure to do so can result in loss of accreditation for the provider, which may then lead to additional negative outcomes, from loss of insurance to a full shutdown by state regulatory agencies.
- U.S. Courts. Federal courts have agreed that healthcare providers have a duty to explain potential risks to patients before treatment since 1972’s Canterbury v. Spence. In 1981, the American Medical Association’s Judicial Council issued an official statement of policy on informed consent. “The patient’s right of self-decision can be effectively exercised only if the patient possesses enough information to enable an intelligent choice,” the Council wrote, as quoted in A History and Theory of Informed Consent. “The patient should make his own determination on treatment.” Physicians who fail to practice informed consent open themselves up to significant legal liability, including malpractice lawsuits.
In short, if a physician breaches informed consent, they will be driven out of practice, either by losing their medical license or because of lawsuits. Informed consent is a necessity for all healthcare providers — although there are a few cases in which the requirement may be waived.
What are some exceptions to the informed consent requirement?
U.S. courts have recognized two general exceptions to the doctrine of informed consent. Other authorities agree with these special cases. A physician may not have to obtain informed consent from a patient if
- An unconscious patient needs immediate attention. “In an emergency situation, informed consent might be waived,” Patwardhan explains. “If the patient is unconscious, and there’s no surrogate there to make the decision or give consent for the patient in an emergency situation, you may go ahead and do the necessary procedure.”
- Discussing the potential treatment is likely to cause serious psychological harm to the patient. If the practice of informed consent itself will harm the patient, the rules change, and a doctrine called “therapeutic privilege” comes into play. According to the Supreme Court of Minnesota’s decision in Cornfeldt v. Tongen (1977), “The therapeutic privilege, a well-recognized exception to the objective standard of disclosure, excuses the withholding of information where disclosure would be unhealthful to the patient.”
Physicians should be careful when applying either of these exceptions to informed consent. When in doubt, always involve the patient in the decision.
According to the AMA Journal of Ethics, “Given that requirements for informed consent are relatively vague and undefined and the exceptions are few, it is in the physician’s best interest to inform patients thoroughly about proposed treatment options, ascertain that they understand their choices, and secure their consent. Doing so will help provide quality patient care and avoid exposure to legal action.”
Who is responsible for obtaining informed consent?
Ultimately, it’s the physician’s responsibility to practice informed consent and ensure that this practice is documented in the patient’s medical record. Any staff member may hand the patient the informed consent form, see that they sign it, and file it. But the doctor who’s treating the patient is the one who’s on the hook for any errors or omissions in the document, or in the conversations through which informed consent is practiced.
For a few real-life examples of who obtains informed consent in various medical settings, read this post. If you’re not sure whether all these informed consent laws apply to your business, continue to Chapter 3.
Who needs informed consent?
The global healthcare industry is enormous, valued at nearly $12 trillion and made up of multiple sectors that, at first glance, don’t seem to have much to do with one another. Veterinarians, big pharma, general practitioners, and insurers are all part of the healthcare industry, but they don’t all practice informed consent all the time. It can be hard to know if you’re in one of the areas of healthcare that’s required to follow these medical consent regulations.
To clear up the confusion, here are a few of the specific fields in which informed consent is an important part of the legal and ethical framework of the profession:
- Clinical healthcare services. Hospitals, urgent care clinics, and doctors’ offices fall into this sector. Physicians who provide clinical care practice informed consent in compliance with medical law, state licensing agencies, and the AMA Code of Ethics, as discussed in the last chapter.
- Mental and behavioral health services. Informed consent in psychology and behavioral health helps keep patients in control of their care. Medical authorities require the practice and documentation of informed consent in mental healthcare, and the American Psychological Association’s Ethical Principles of Psychologists and Code of Conduct states that “when psychologists conduct research or provide assessment, therapy, counseling, or consulting services…they obtain the informed consent…using language that is reasonably understandable…” The APA’s Ethical Principles also require psychologists to “appropriately document written or oral consent, permission, or assent.” Learn more about informed consent in psychology here.
- Research involving human subjects. Psychological studies, drug tests, medical device trials, and healthcare research programs often involve human subjects. In these instances, researchers must obtain informed consent from each participant to include them in the study. In the U.S., the Food and Drug Administration, the Department of Health and Human Services, and local institutional review boards require this informed consent, and monitor researchers to make sure their documentation is in order. Learn more about informed consent in research, specifically for medical device approval, here.
The essential rule is that you must practice informed consent if your business treats or studies human beings. You must also document the patient’s agreement with a signed form. While the healthcare industry is unique in its informed consent requirements, some other service providers have started using the term as well.
Informed consent outside the healthcare industry
In the wake of COVID-19, more business owners asked customers to sign health waivers in order to access services. The consent form has become a mainstay at virtually any business or organization that places people close to one another: barbershops, salons, massage studios, gyms, co-working spaces, restaurants, daycare providers, health clubs, schools, and more.
Businesses collected these consent forms to protect themselves from liability. If a customer contracted COVID-19 after a trip to the salon, the hairstylists didn’t want to end up on the wrong side of a lawsuit.
Some employers call these liability waivers “informed consent” documents, but that’s not entirely accurate. Sometimes they mirror the elements of a medical informed consent form, listing potential risks and benefits of the service, even describing the customer’s confidentiality rights. But it’s important to distinguish between “informed consent” as a legal term and its use as a simple descriptor. In the healthcare industry, informed consent is required by law; that’s not the case at a beauty salon.
So if you run a small business, should you ask customers to sign a health waiver? It’s not a bad idea if you’re worried about frivolous lawsuits, lawyer Bettina Strauss told Consumer Reports in July 2020. “A waiver doesn’t prevent people from filing lawsuits, but it’s a disincentive for a lawyer to take a case on and it would allow a company a quicker exit from a frivolous lawsuit should one be filed,” Strauss said.
If you need an online consent form, in or out of the healthcare field, Jotform can help. In the final chapter of this guide, we’ll explain how.
Informed consent forms
Physicians must “document the informed consent conversation and the patient’s (or surrogate’s) decision in the medical record,” says the AMA’s Code of Medical Ethics. The FDA makes similar demands on researchers and their subjects. In both cases, informed consent forms do the job.
We discussed the elements of informed consent in Chapter 1. But which elements actually have to be listed on the form? After all, the Joint Commission describes informed consent as “a process of communication.” Some of the communication is verbal, and not all the information must appear on the page — at least not in clinical practice (we’ll get to the special case of research shortly).
One influential study published in the Archives of Surgery recommends four basic elements that physicians should include on their informed consent forms, regardless of the conversation in the examination room:
- The nature of the medical procedure
- Risks associated with the treatment
- Projected benefits
- Alternative treatments
Many physicians also include other information on informed consent forms: confidentiality rights under HIPAA; identifying information of all doctors involved in the treatment (surgeons, anesthesiologists, etc.); hospital policies, terms, and conditions; and other legal details, according to state or local statutes. Informed consent forms will differ from one medical sector to the next. For example, compare this professional counseling informed consent form with this one designed for chiropractors.
In the FDA-dominated world of pharmaceutical research, informed consent documents are a bit more regimented and a lot more comprehensive.
Requirements for informed consent forms in pharmaceutical research
The FDA is very clear about the information researchers must include in their informed consent documentation. The administration lists eight elements that researchers must include on their informed consent forms:
- An overview of the study
- Possible hazards of participation
- Potential benefits of participation
- Other medical treatments for the condition under study
- A description of the patient’s confidentiality rights
- Compensation, plus how researchers plan to treat study-related injuries/illnesses
- Contact information for authorities involved in the study
- A statement reiterating that the subject’s participation is totally voluntary and can be withdrawn at any time
Researchers may include even more information on their informed consent forms “when appropriate,” says the FDA. They might need to mention that there could be hidden risks to participation, for instance, or what could happen if the subject suddenly quits the study.
Electronic informed consent forms from Jotform
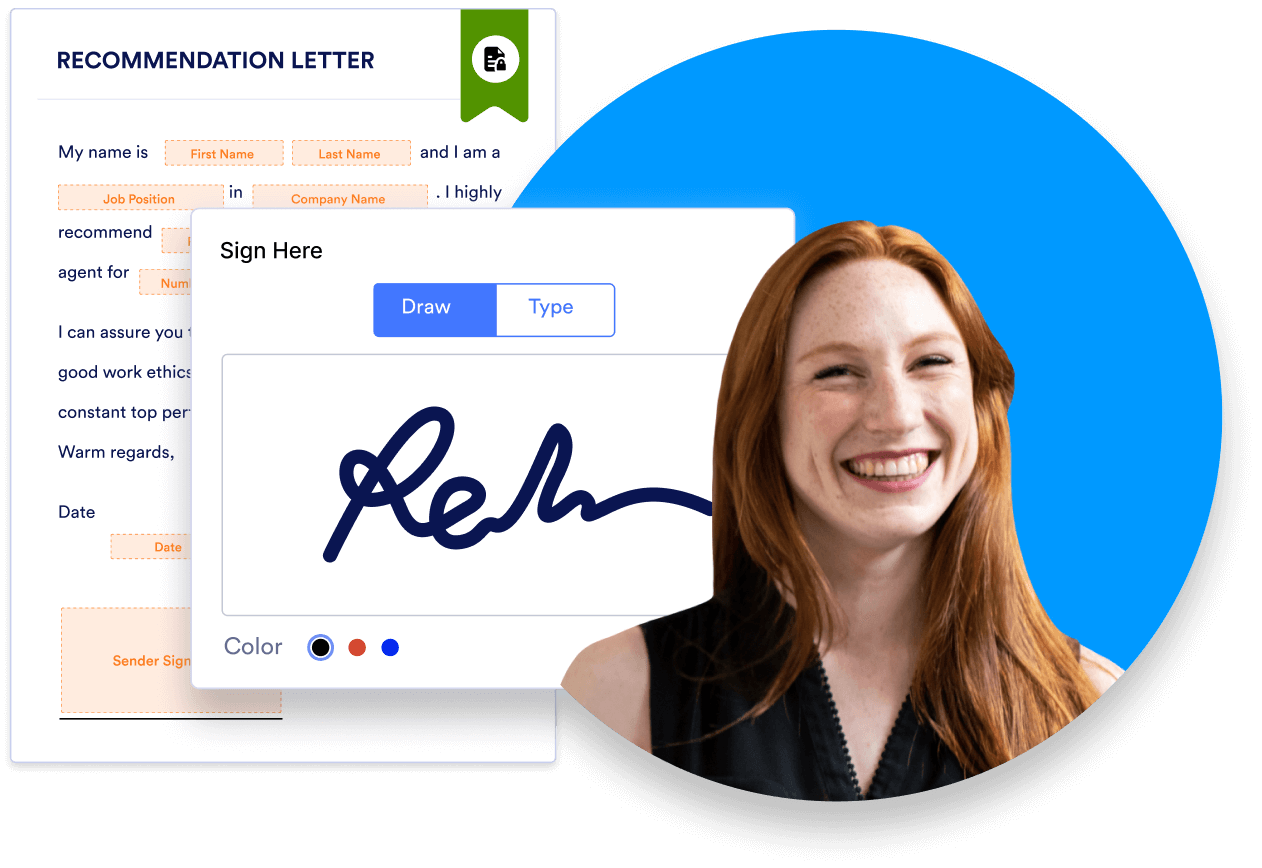
By now, it’s clear that informed consent documents can get long and complicated. That only adds to the challenge of sharing, collecting, and filing them. One solution is to switch to online forms, which save paper and can be transmitted electronically. Because they’re already digital, staff can easily add online signed consent forms to electronic medical record systems — no scanning necessary.
Jotform makes it easy to personalize, deploy, sign, and collect HIPAA-enabled electronic consent forms. Start with one of our 60+ informed consent templates, and edit to fit your situation. Jotform’s intuitive drag-and-drop editor lets you add and remove sections at will. Just select a field to replace the template text with your own copy.
You can use e-signature widgets from Adobe Sign or DocuSign, or use Jotform’s E-signature widget. Recipients can sign your forms on virtually any device, including Android and iOS mobile platforms. Learn more about e-signatures from Jotform here, or choose a HIPAA-enabled Gold plan to start building online informed consent forms today.








Send Comment: